
Master Cleaning Validation Plan
LOCATION : ===


COMPANY NAME :
ADDRESS
1.0 APPROVAL OF MASTER CLEANING VALIDATION PLAN
This Master Cleaning Validation Plan is a master document, which describes the entire scope, procedure, consideration and details of the cleaning validation activities, to be carried out at the dosage form manufacturing facility of XXXXXXXXXXXXXXXXXXXX.
This Master Cleaning Validation Plan has been approved by the following:


Table of Contents
1.0 Approval of Master Cleaning Validation Plan
2.0 Objective
3.0 Scope & Validation Policy
4.0 Responsibility for Cleaning Validation activities
4.1 Validation Team Members
5.0 Introduction
6.0 Cleaning and Extent of Cleaning
6.1 Drug Products / Formulations
6.2 Type A Cleaning
6.3 Type B Cleaning
7.0 Development of Cleaning Validation Program
7.1 Identification of Potential Contaminants
7.2 Product Grouping and Worst Case Selection
7.3Manufacturing Equipment Grouping and Worst Case Selection
7.4 Selection of Cleaning Procedure
7.4.1 Automated Cleaning Procedure
7.4.2 Manual Cleaning Procedure
7.5 Selection of Sampling Method
7.5.1 Swab (Direct Surface) Sampling Method
7.5.2 Rinse (Wash Water) Sampling Method
7.6 Selection of Analytical Method
7.6.1 Specific Analytical Method
7.6.2 Non-Specific Analytical Method
7.7 Analytical Method Validation
7.7.1 Limit of Detection
7.7.2 Limit of Quantification
7.8 Determination of Recovery Factor and Limit
7.9Establishment of Limits and Acceptance Criteria
7.9.1Residue of the Previously Manufactured Product
7.9.2 Residue of Cleaning Agent
7.9.3 Microbial Consideration
8.0 Cleaning Validation Strategy
8.1Data Interpretation and Drawing Conclusion
8.2 Step by step Approach of Cleaning Validation
9.0 Ongoing Verification of Cleaning
9.1 Visual Inspection
9.2 Verification by Testing
10.0 Revalidation
10.1Revalidation due to Change
10.2Periodic/ Routine Revalidation
11.0Documentation for Cleaning Validation
11.1CleaningValidation Protocol
11.2 Cleaning Validation Report
12.0 Annexures
OBJECTIVE
The objective of the Master Cleaning Validation Plan (MCVP) is to provide a plan for development of cleaning validation procedure and revalidation program for pharmaceutical equipment's that are not only in line with the cGMP and current regulatory requirements but are also practical, cost effective and based on good science.
MCVP adds value to our quality system and provides assurance on product safety, by ensuring proper cleaning of processing equipment and utensils remain in a state of validation.
3.0 SCOPE & VALIDATION POLICY
This MCVP is applicable for cleaning and cleaning validation at manufacturing location of XXXXXXXXXXXX. This document on Cleaning Validation is intended to address special considerations and issues pertaining to validation of cleaning procedures for equipment used in the manufacture of pharmaceutical dosage forms. The document is also intended to establish consistency and uniformity with respect to equipment cleaning procedures. This MCVP is intended to cover validation of equipment cleaning for the removal of contaminants associated with previous products, residues of cleaning agents as well as the control of potential microbial contaminants.
The cleaning validation program covers all the manufacturing operations, which are listed below:
§ XXXXX
§ XXXX
§ XXXXX
§ XXXX
4.0 RESPONSIBILITIES FOR CLEANING VALIDATION ACTIVITIES
The cleaning validation activities shall be carried out by team of Personnel from Validation / Quality Assurance, Quality Control, Production and Engineering department, as applicable. The respective department head shall ensure that the person nominated for the cleaning validation activity is having adequate knowledge and experience in carrying out the activity / task.
The Quality Assurance / Validation department shall be responsible for coordination of the cleaning validation activities, facilitating the protocol preparation, execution of cleaning validation activities and compilation of reports of cleaning validation activities related to manufacturing equipment's and utensils.
Quality control department shall be responsible for determination of recovery factors by spiking, analytical method development and it’s validation. Quality control department shall also provide support for the analysis of the samples collected during the cleaning validation activities.
The respective production department shall prepare and review the cleaning validation protocols related to their respective areas, execute the cleaning validation activities and compile the validation reports.
Engineering department shall provide all necessary support for the execution of cleaning validation by maintaining processing equipments in the state of repair and providing the product contact surface areas.
Quality assurance department shall review & approve the cleaning validation protocols before implementation, monitor the progress of the cleaning validation activities and approve the compiled Validation reports. The final approval of validation reports shall be done by Quality Assurance.4.1 Validation Team Members
The Cleaning validation activity shall be coordinated by a validation team, which shall comprise of a working group of qualified personnel, as required.The Cleaning Validation Team may be made up of representatives from the following areas:
§ Production
§ Quality Control
§ Engineering
§ Quality Assurance
Other personnel may be co-opted, as and when necessary, as members of the cleaning validation team, to assist during the cleaning validation on manufacturing equipments, and utensils.
The Cleaning Validation Team shall be responsible for :
§ Preparation of Master Cleaning Validation Plan.
§ Preparation product and equipment matrix.
§ Setting up acceptance criterion (Chemical and Microbiological).
§ Identifying the equipments and utensils on which cleaning validation shall be performed.
§ Facilitating the preparation of cleaning validation protocols and reports.
§ Determining the suitability of established protocols, to validate the cleaning procedures.
§ Verifying the adequacy of tests used for the cleaning verification, as established by the cleaning validation protocol.
§ Approving all formats, to be used for the reporting of test data during cleaning validation.
§ Reviewing each protocol contents, to assure compliance with current regulations and guidelines.
§ Approving the original protocols and final validation data for each cleaning validation.
§ Determine the extent of re-validation necessary in case of changes to a validated system.
§ Training for the personnel, related to cleaning validation activities
5.0 INTRODUCTION
The objective of Master Cleaning Validation Plan of Equipment, utensils and components is to establish sufficient documented evidence to assure that the cleaning procedures can reproducibly remove residue of the product being handled below established acceptance limit. The quantity of residue of the previous product if, within the acceptance limit will not affect quality and Safety of the subsequent product to be manufactured by using same equipment and facility.
The objective of Cleaning Validation is to establish cleaning procedures & residue limits that are practical, achievable, and verifiable and assure safety. Cleaning of the equipment train, individual equipment, utensil and / or components are carried out separately and clubbed with a pre-wash and inspection program. The cleaning procedure comprise of thorough cleaning with Sodium Lauryl Sulphate and soft water in suitable combination followed by final rinsing with Purified Water.
During development of cleaning validation plan and protocols due importance should be paid to the residue and contaminants. The residue and contaminants shall include
§ Desired actives (of previously manufactured product)
§ Microbes
§ Equipment related materials such as equipment linings, gaskets, filter agents and / or lubricants.
6.0 CLEANING AND EXTENT OF CLEANING
Drug Products / Formulations
In manufacturing process, two different cleaning situations arise and each situation are described as follows;
* Cleaning between batch-to-batch changeover.
* Cleaning between product-to-product changeover.
In our non-dedicated drug product manufacturing facility different cleaning procedures are developed depending upon the change over situations.
6.1.1 Type A Cleaning
Used only when cleaning between different batches of the same product; i.e. batch to batch cleaning.
Example – In manufacturing of product X, there are 2 Batches to be manufactured one after another as shown in the following flow chart.
Batch A of Product X ---------> Batch B of Product X
For a particular equipment and / or equipment train, if batch A is to be followed by Batch B, then a type A cleaning is required.
6.1.2 Type B Cleaning
Used when cleaning between different Product-Batches; i.e. product to product cleaning.
Example – In manufacturing of product X followed by product Y as shown in the following flow diagrams.
Batch A of Product X ---------> Batch B of Product Y
The two different types of cleaning (Level A and Level B) differ in thoroughness of the cleaning process, cleaning conditions, the level of verification of cleaning and degree of risk associated with contamination, as shown in the following order.

7.0 DEVELOPMENT OF CLEANING VALIDATION PROGRAM
Only B cleaning is subjected to be validated. A systematic cleaning validation program shall be followed which are described in the following sub-sections.
7.1 Identification of Potential Contaminants
The contaminants in pharmaceutical product manufacturing may be of different types like the active substances of previously manufactured drug product, cleaning agents, microorganisms, lubricants, solvents, equipment related, etc. Among these the potential contaminants are active of previously manufactured drug product and microorganisms, which shall be considered in cleaning validation.
7.2 Product Grouping and Worst Case Selection
During development of cleaning validation program the products shall be grouped to identify that particular product which will represent the worst case and making the cleaning validation program manageable. The philosophy behind product grouping is as follows:
§ All products having same dosage form. e.g. XXXXXXXXXXXXXX etc.
§ All products having same active ingredients.
All products shall be listed in the form of a matrix, showing corresponding batch size, dosage strength, amount of active ingredients, minimum daily dose (of active), maximum daily dose (of drug product), water solubility, preferable swab medium and step by step calculated results. The product(s) that shows the highest dosage strength and lowest acceptance criteria with respect to less water solubility can be considered as the product of worst case. It is common that practically the perfect worst case may not come in a single product; in that case more than one product shall be selected as worst case. Bracketing concept can be used to show the extremity of cleaning procedure.
7.3 Manufacturing Equipment Grouping and Worst Case Selection
All processing equipment's shall be arranged in train as per the manufacturing flow of product. This comes right from the dispensing of raw materials to primary packaging.
The equipment train having highest product contact surface area shall be considered as worst case.
7.4 Selection of Cleaning Procedure
Same cleaning procedure shall be followed for all the manufacturing equipments and utensils irrespective of the product. There are following two types of cleaning procedures used in our manufacturing facility.
7.4.1 Automated Cleaning Procedure
§ Cleaning of the equipment is performed in place without disassembling.
§ Cleaning process may be controlled by an automated program.
§ Very consistent in nature.
7.4.2 Manual Cleaning Procedure
§ Cleaning of the disassembled equipment is performed
§ Effectiveness largely depends upon the person performing the cleaning operation.
For any type of cleaning method used, cleaning validation is a basic requirement, samples shall be taken, analyzed and results documented.
After Validation of the cleaning method, suitable reports shall be prepared to demonstrate that critical parameters such as the temperature, cycle time & sequence, quantity of detergent used etc. are still being achieved and / or followed on a routine basis. These records shall be incorporated in the Cleaning Validation Report.
Once the cleaning method is validated, it shall be ensured that the method is not changed. If required any change in the validated cleaning method shall be governed as per the company’s existing change control policy and the need for re-validation shall be evaluated.
7.5 Selection of Sampling Method
There are two kind of sampling method shall be followed during validation; e.g. Swab (Direct Surface) Sampling method and Rinse (Wash Water) Sampling Method. Direct surface monitoring should be used for validation as well as regular cleaning verification. All samples shall adequately be identified by their identification numbers, provided in the individual cleaning validation protocols.
7.5.1 Swab (Direct Surface) Sampling Method
§ Absorbent cotton swab of 5 cm. × 5 cm. (25 cm2) shall be used.
§ Residue shall be collected by physical rubbing of swab on the cleaned equipment / utensil surface.
§ The swab shall be wet by the solvent, specified in the preferable swab medium column in the product matrix.
§ Finally the sampled swab shall be desorbed in the glass bottle containing 20 ml. of that specific solvent.
§ Most difficult to clean locations shall be selected for swab sampling.
§ It is recommended to collect swabs at least from 5 locations (in case of swab taken for chemical residue) of each major processing equipment.
§ The sampling locations along with the drawing shall be specified in the cleaning validation protocol(s).
§ Readymade Sterile swab shall be used for microbiological sampling.
7.5.2 Rinse (Wash Water) Sampling Method
§ Large surface area can be easily sampled by this method.
§ Strongest preferred method for difficult to access parts of equipment.
§ Rinse sample shall be collected in conjunction with swab samples.
§ 2 liters of purified water, representing total equipment (product contact area) surface area shall be used for final rinsing.
§ Finally 100 ml. of water shall be collected in glass bottle as rinse sample.
Details of sampling procedure shall be described in individual cleaning validation protocols.
7.6 Selection of Analytical Method
It is important to select an appropriate method for detection of the residue in the cleaning sample. Analytical method shall be selected as per the applicability depends on the residue of the active to be analyzed. Methods shall be validated for accuracy, precision, linearity, ruggedness, robustness, sensitivity and recovery.
7.0 DEVELOPMENT OF CLEANING VALIDATION PROGRAM
Only B cleaning is subjected to be validated. A systematic cleaning validation program shall be followed which are described in the following sub-sections.
7.1 Identification of Potential Contaminants
The contaminants in pharmaceutical product manufacturing may be of different types like the active substances of previously manufactured drug product, cleaning agents, microorganisms, lubricants, solvents, equipment related, etc. Among these the potential contaminants are active of previously manufactured drug product and microorganisms, which shall be considered in cleaning validation.
7.2 Product Grouping and Worst Case Selection
During development of cleaning validation program the products shall be grouped to identify that particular product which will represent the worst case and making the cleaning validation program manageable. The philosophy behind product grouping is as follows:
§ All products having same dosage form. e.g. XXXXXXXXXXXXXX etc.
§ All products having same active ingredients.
All products shall be listed in the form of a matrix, showing corresponding batch size, dosage strength, amount of active ingredients, minimum daily dose (of active), maximum daily dose (of drug product), water solubility, preferable swab medium and step by step calculated results. The product(s) that shows the highest dosage strength and lowest acceptance criteria with respect to less water solubility can be considered as the product of worst case. It is common that practically the perfect worst case may not come in a single product; in that case more than one product shall be selected as worst case. Bracketing concept can be used to show the extremity of cleaning procedure.
7.3 Manufacturing Equipment Grouping and Worst Case Selection
All processing equipment's shall be arranged in train as per the manufacturing flow of product. This comes right from the dispensing of raw materials to primary packaging.
The equipment train having highest product contact surface area shall be considered as worst case.
7.4 Selection of Cleaning Procedure
Same cleaning procedure shall be followed for all the manufacturing equipments and utensils irrespective of the product. There are following two types of cleaning procedures used in our manufacturing facility.
7.4.1 Automated Cleaning Procedure
§ Cleaning of the equipment is performed in place without disassembling.
§ Cleaning process may be controlled by an automated program.
§ Very consistent in nature.
7.4.2 Manual Cleaning Procedure
§ Cleaning of the disassembled equipment is performed
§ Effectiveness largely depends upon the person performing the cleaning operation.
For any type of cleaning method used, cleaning validation is a basic requirement, samples shall be taken, analyzed and results documented.
After Validation of the cleaning method, suitable reports shall be prepared to demonstrate that critical parameters such as the temperature, cycle time & sequence, quantity of detergent used etc. are still being achieved and / or followed on a routine basis. These records shall be incorporated in the Cleaning Validation Report.
Once the cleaning method is validated, it shall be ensured that the method is not changed. If required any change in the validated cleaning method shall be governed as per the company’s existing change control policy and the need for re-validation shall be evaluated.
7.5 Selection of Sampling Method
There are two kind of sampling method shall be followed during validation; e.g. Swab (Direct Surface) Sampling method and Rinse (Wash Water) Sampling Method. Direct surface monitoring should be used for validation as well as regular cleaning verification. All samples shall adequately be identified by their identification numbers, provided in the individual cleaning validation protocols.
7.5.1 Swab (Direct Surface) Sampling Method
§ Absorbent cotton swab of 5 cm. × 5 cm. (25 cm2) shall be used.
§ Residue shall be collected by physical rubbing of swab on the cleaned equipment / utensil surface.
§ The swab shall be wet by the solvent, specified in the preferable swab medium column in the product matrix.
§ Finally the sampled swab shall be desorbed in the glass bottle containing 20 ml. of that specific solvent.
§ Most difficult to clean locations shall be selected for swab sampling.
§ It is recommended to collect swabs at least from 5 locations (in case of swab taken for chemical residue) of each major processing equipment.
§ The sampling locations along with the drawing shall be specified in the cleaning validation protocol(s).
§ Readymade Sterile swab shall be used for microbiological sampling.
7.5.2 Rinse (Wash Water) Sampling Method
§ Large surface area can be easily sampled by this method.
§ Strongest preferred method for difficult to access parts of equipment.
§ Rinse sample shall be collected in conjunction with swab samples.
§ 2 liters of purified water, representing total equipment (product contact area) surface area shall be used for final rinsing.
§ Finally 100 ml. of water shall be collected in glass bottle as rinse sample.
Details of sampling procedure shall be described in individual cleaning validation protocols.
7.6 Selection of Analytical Method
It is important to select an appropriate method for detection of the residue in the cleaning sample. Analytical method shall be selected as per the applicability depends on the residue of the active to be analyzed. Methods shall be validated for accuracy, precision, linearity, ruggedness, robustness, sensitivity and recovery.
7.6.1 Specific Analytical Test Methods
Specific analytical test methods shall be applicable for identification and quantitative analysis of microbes / active residue in swab / rinse samples. Some common examples of specific analytical methods are given;
§ High Performance Liquid Chromatography (HPLC).
§ Ultra violate Spectrophotometer (UV).
§ Microbial Test.
7.6.2 Non-Specific Analytical Methods
Although it is expected to follow specific analytical test methods for the cleaning samples, but many of the non-specific methods are preferable because these are simple, fast and ability to provide overall contamination level of cleaning. Some examples are mentioned below;
§ Visual Examination
§ pH
§ Total Organic Carbon (TOC)
§ Conductivity
Non-specific analytical methods shall be utilized to analyze the rinse water sample during cleaning validation / cleaning verification.
7.7 Analytical Method Validation
The analytical method used for evaluation of cleaning efficacy shall be validated prior execution.
7.7.1 Limit of detection (LOD)
Limit of Detection is the lowest amount of the analyte in the sample that can be detected qualitatively. The analytical method should be sensitive enough to detect the active residue present in the sample. The limit of detection shall be established during the validation of analytical method.
7.7.2 Limit of Quantification (LOQ)
Limit of Quantification is the lowest amount of the analyte in the sample that can be detected qualitatively. The analytical method shall be validated not on the basis of L2* but rather on the L3* basis. This fourfold factor in limit of quantification is significant for successful cleaning validation.
7.8 Determination of Recovery Factor and Limit
The recovery studies shall be a part of cleaning validation studies. The limit L3 is the acceptance limit in the analyzed sample. It shall be adjusted by swab recovery factor and the ultimate outcome will be L4*.
Typically a recovery study comprises of the following:
§ Deliberately contaminating the subjected equipment surface and / or similar coupon surface (predefined area) with known concentration of the molecule of interest.
§ The interested molecule residue shall be recovered form the same surface by using the same sampling techniques (rinse / swab), which would be used during the validation studies.
§ The recovered quantity shall be determined by using the same validated analytical methods, which would be used during the validation studies.
§ A recovery factor shall be established based on statistical evaluation of the results.
§ The following table may be used for determination of recovery factor(s);
Specific analytical test methods shall be applicable for identification and quantitative analysis of microbes / active residue in swab / rinse samples. Some common examples of specific analytical methods are given;
§ High Performance Liquid Chromatography (HPLC).
§ Ultra violate Spectrophotometer (UV).
§ Microbial Test.
7.6.2 Non-Specific Analytical Methods
Although it is expected to follow specific analytical test methods for the cleaning samples, but many of the non-specific methods are preferable because these are simple, fast and ability to provide overall contamination level of cleaning. Some examples are mentioned below;
§ Visual Examination
§ pH
§ Total Organic Carbon (TOC)
§ Conductivity
Non-specific analytical methods shall be utilized to analyze the rinse water sample during cleaning validation / cleaning verification.
7.7 Analytical Method Validation
The analytical method used for evaluation of cleaning efficacy shall be validated prior execution.
7.7.1 Limit of detection (LOD)
Limit of Detection is the lowest amount of the analyte in the sample that can be detected qualitatively. The analytical method should be sensitive enough to detect the active residue present in the sample. The limit of detection shall be established during the validation of analytical method.
7.7.2 Limit of Quantification (LOQ)
Limit of Quantification is the lowest amount of the analyte in the sample that can be detected qualitatively. The analytical method shall be validated not on the basis of L2* but rather on the L3* basis. This fourfold factor in limit of quantification is significant for successful cleaning validation.
7.8 Determination of Recovery Factor and Limit
The recovery studies shall be a part of cleaning validation studies. The limit L3 is the acceptance limit in the analyzed sample. It shall be adjusted by swab recovery factor and the ultimate outcome will be L4*.
Typically a recovery study comprises of the following:
§ Deliberately contaminating the subjected equipment surface and / or similar coupon surface (predefined area) with known concentration of the molecule of interest.
§ The interested molecule residue shall be recovered form the same surface by using the same sampling techniques (rinse / swab), which would be used during the validation studies.
§ The recovered quantity shall be determined by using the same validated analytical methods, which would be used during the validation studies.
§ A recovery factor shall be established based on statistical evaluation of the results.
§ The following table may be used for determination of recovery factor(s);

7.9 Establishment of Limits and Acceptance Criteria
The equipment and utensil surface are should be visually clean. The rationale for the residue limit shall be scientific, logical and based upon the knowledge of the material. It shall also be practical, achievable and verifiable. The quantitative acceptance criteria shall be considered as the best for the determination of residue limits. In cleaning validation, two types of residues and microbiological consideration shall be of prime importance as these have major impact on equipment cleanliness and processing product quality. These residues and their acceptance limits are as follows;
7.9.1 Residue of the previously manufactured product
Mainly the active ingredient(s) are concern. The acceptance criterion shall be based on the therapeutic dose and safety factor of the active being used in the previously manufactured drug product at the same equipment.
§ Safety factor is the allowing not more than a fraction of a therapeutic dose to be present in the subsequent product. The safety factors may vary, as the degree of risks is different for different dosage forms.

§ Limit Calculation On The Basis Of Therapeutic Dose
The step-by-step approach shall be followed for the determination of limit. The following formula shall be used;
Step – 1
Purpose: To calculate the limit of active agent in any subsequently manufactured product (LSP).
Information required:
(i) Minimum daily dose of the active being cleaned (TD).
(ii) Maximum daily dose of the subsequently manufactured drug product (LDD). (iii) Safety factor of the active being cleaned (SF).
Formula: LSP=SF × TD/LOD
Step – 2
Purpose: To determine the residue limit of active contamination level per area of equipment (L2*).
Information required:
(i) LSP from 1st stage.
(ii) The batch size of the subsequent product (BSSP in kg.)
(iii) The shared equipment surface area (SESA in cm2).
Formula:L2 = LSP × BSSP X 100 /SESA
Step – 3
Purpose: To determine the residue limit for the analytical sample (L3*).
Information required:
(i) The surface area residue limit (L2).
(ii) The surface area swabbed (in cm2).
(iii) The amount of solvent the swab is desorbed (in gm.).
Formula: L3= L2 × Swabbed surface area / Amt. of desorption solvent
Step – 4
Purpose: To determine the residue limit for the analytical sample considering swab recovery factor (L4*).
Information required:
(i) Residue limit for the analytical sample (L3).
(ii) Swab recovery factor determined by practical experiment.
Formula: L4 = L3/ Swab Recovery Factor
§ Limit Determination On The Basis Of 10 ppm Criterion
As per cGMP, the highest allowable residue of active should not exceed 10 ppm level. On that basis the 10 ppm limit shall be considered conservatively in place of the dosage criterion limit on those case(s) where calculated limit will be greater than 10 ppm.
7.9.2 Residue of Cleaning Agent
The cleaning program shall be designed to demonstrate the removal of cleaning agent below the acceptance level. As per the established cleaning procedure, sodium lauryl sulphate is used to clean the processing equipments and utensils. Not more than 10 ppm of the detergent residue shall be considered as acceptance criteria.
7.9.3 Microbiological Consideration
Sufficient documented evidence shall be established to demonstrate that the routine cleaning and storage of the cleaned equipment does not allow microbial growth, beyond the defined limit. Any equipment cleaned shall be dried and protected to ensure that, there will be no microbial proliferation and microbial contamination of the cleaned equipment.
Practically it cannot expect equipment free from all microorganisms. Setting of the microbial limit is important and required because even a single organism in the equipment could possibly result in significantly higher contamination level in the product manufactured using that equipment.
Three kind of microbiological limit can be categorized;
§ The first criteria with respect to the microbial contamination limit of the equipment surface, is absence of pathogenic organisms such as E. coli, Enterococcus, Salmonella, etc (commonly known as USP indicator organisms).
§ Not more than 10 cfu / 25 cm2 is recommended as swab limit.
§ Rinse samples shall not be exceeding 100 cfu / ml.
8.0 CLEANING VALIDATION STRATEGY
§ The manufacturing equipments shall be cleaned as per the preestablished Standard Operating Procedures (SOPs) on equipment cleaning.
§ The whole cleaning validation study shall be divided into two phases; first phase is the Introductory Phase and second phase is Confirmatory Phase.
§ In the Introductory Phase, three consecutive batches of considered product(s) shall be taken for cleaning validation study in which 100 % swab sampling shall be done in conjunction with rinse water collections.
§In the Confirmatory Phase, only rinse water sampling shall be conducted for next three batches of same product(s).
8.1 Data Interpretation and Drawing Conclusion
§ All test results shall be verified in accordance with the established acceptance criteria.
§ Data generated during cleaning validation shall be systematically documented to facilitate sound understanding regarding the effectiveness of existing cleaning methods.
§ Analytical results complying the established acceptance criteria shall be considered as the root of a successful cleaning validation study.
§ A correlation shall be estimated in between the result obtained from swab sample analysis and result obtained from rinse water analysis.
§ Equipment wise, comparative graphical representation of analytical test results can be used as a tool of showing relation between two types of samples.
§ The test results of rinse water samples of next three batches shall be served as a confirmatory evaluation of the cleaning efficacy. In these cases, the analytical results should not give rise an unexpected result.
§ If the out comes of all six batches are consistent with the predetermined limits, the cleaning validation can be considered as successful.
§ Any deviation or nonconformity of the cleaning process, sampling method, analytical method and test results, shall be fully investigated and decision may be taken for further consideration of cleaning validation.
9.0 ONGOING VERIFICATION OF CLEANING
Verification of cleaning shall be conducted regularly after completion of cleaning validation. It involves the performance of visual inspection of cleaned equipments and / utensils and testing of collected samples. However additional verification may be necessary depending on the complexity of the equipment / product being cleaned.
9.1 Visual Verification
The cGMP regulations require visual inspection of each piece of equipment immediately before use to ensure its cleanliness. No traces of contaminants should remain on the equipment and utensil surface. After successful completion of cleaning validation (for both Introductory Stage and Confirmatory stage), visual inspection shall be considered as vital tool for verification of cleaning effectiveness. The visual inspection procedure is mentioned bellow;
The visual inspection is subjected to follow for each and every batch of product being cleaned.
It is applicable for both types of cleaning A and B.
Cleaned equipment surfaces shall be inspected with naked eye at a distance of approximately 25 cm.
Adequate quantity and intensity of light should be present in the area during visual inspection.
Visual inspection shall be conducted just before the use of cleaned equipments / utensils for processing of next batch / product.
The results of visual inspection shall be documented in the respective BMR / BPR.
Any deviation from the anticipated result shall be mentioned in the respective BMR / BPR and subjected to investigation.
9.2 Verification by Testing
The validated cleaning method shall be verified with analytical testing during periodic revalidation. The procedure of verification by testing is described as follows;
Rinse (Wash Water) shall be considered as the preferred sampling method for cleaning verification.
Rinse water shall be collected as per the method described under the heading of “ 7.5.2 Rinse (Wash Water) Sampling Method ” in earlier discussion.
Non-Specific Analytical Testing Methods (e.g. TOC, Conductivity, pH) shall be used for the quantitative analysis of residue.
Test results shall be documented in accordance with acceptance criteria in the periodic revalidation report followed by conclusion.
Any deviation from the anticipated result shall be mentioned in the respective cleaning validation protocol and subjected to investigation.
10.0 REVALIDATION
Revalidation shall be conducted to demonstrate that the existing cleaning procedure is in the state of validation. There are two different types of Cleaning Revalidation can be conducted depending upon the situation arises and consideration of the cleaning validation team members.
10.1 Revalidation due to Change
The degree of revalidation due to Change should be based on the risk assessment of the change / observation regarding the cleaning efficacy. The list of changes is mentioned below which may affect the existing cleaning validation status.
Modification in the existing equipment design.
Changes in the equipment / utensil cleaning method.
Changes in the analytical sample testing method.
Changes in detergent used for cleaning.
Changes in sampling method / tool regarding cleaning.
New product in the facility that challenges the existing worst-case consideration.
Addition of new equipment(s) in the processing line which increases the total surface area of entire equipment train and challenges the pre-considered surface area of largest equipment train.
Revalidation due to change can be partial validation / full validation depending upon the situation on which revalidation will be considered. It is the responsibility of cleaning validation team to determine the effect of proposed changes on the current state of cleaning. The major changes in the existing equipments, critical cleaning procedure and analytical method shall be subjected to go through the company’s change control procedure. A new cleaning validation protocol shall be prepared, followed by preparation of report.
10.2 Periodic / Routine Revalidation
Periodic or Routine Cleaning Revalidation shall be performed to demonstrate that the previously validated cleaning procedure remains in the state of validation during a specified time period.
Each periodic revalidation shall be conducted after one year from the date of completion of previous validation / revalidation study.
At this time, validation protocol shall not be required but report shall be prepared for each revalidation study.
The study pattern of periodic revalidation has already been mentioned earlier under the heading of “ 9.2 Verification by Testing”.
Any deviation observed during revalidation shall be documented in the cleaning revalidation report.
11.0 DOCUMENTATION FOR CLEANING VALIDATION
Further documentation shall be prepared for each cleaning validation study, which will be product specific. The documentation for cleaning validation comprises of Protocol and Report. The details of protocol and report are specified as follows;
11.1 Cleaning Validation Protocol
An authorized product specific validation protocol shall be prepared prior to conducting the cleaning validation study. The product representing the worst case shall be subjected to get validated. The cleaning validation protocol shall contain the following heads but may not be limited to this;
Objective
Scope
Description of the process
Identification of critical monitoring parameter (for the cleaning procedure).
Test Functions (Tests to be utilized for validation of cleaning procedure).
Sampling Locations and Procedures (with relevant drawing).
Analytical Test Methods
Limit and Acceptance Criteria
11.2 Cleaning Validation Report
Cleaning Validation Report shall be prepared in accordance with the established protocol. It may contain the following fields;
Inprocess Observations and Visual Inspection results.
Analytical Observations and results.
Data interpretations and statistical evaluation of results.
Relationship of test results between swab and rinse sample analysis.
Any Deviations Observed.
Conclusion.
12.0 ANNEXURES
These are the supporting documents, which are the integral part of this Master Cleaning Validation Plan. All annexure(s) are subjects of getting updated frequently as per the requirement. The update of annexure(s) shall not affect the revision status of the Master Cleaning Validation Plan (MCVP).
The list of annexure(s) are given below;
Product Matrix.
Equipment Matrix.
List of SOPs related to cleaning of processing equipment's and utensils.
PRODUCT MATRIX
|
Date of Revision |
|
EQUIPMENT MATRIX
|
Date of Revision |
|
LIST OF SOPs RELATED TO CLEANING OF PROCESSING EQUIPMENTS AND
UTENSILS
|
Date of Revision |
|


.JPG)













![Difference between Stability[Shelf life] Specification and Release Specification](https://4.bp.blogspot.com/-O3EpVMWcoKw/WxY6-6I4--I/AAAAAAAAB2s/KzC0FqUQtkMdw7VzT6oOR_8vbZO6EJc-ACK4BGAYYCw/w680/nth.png)
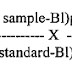
){kind=link}
0 Comments