
1.0 OBJECTIVE
To lay down the procedure for Validation of Analytical test method for Dissolution by UV.
2.0 SCOPE
This SOP is applicable for validation of Analytical test method for dissolution by UV in Analytical Research department at Company Name.
3.0 BACKGROUND
NIL
4.0 RESPONSIBILITY
4.1 Analytical research personnel whoever is operating the Equipment.
4.2 Group Leader Analytical research to ensure proper and safe operation of the Equipment.
4.3 Head - Analytical research or his designee to ensure overall compliance
5.0 PROCEDURE

5.1 Linearity of detector response:
5.1.1 Demonstrate the linearity of detector response in the range of 25% to 150% of the target concentration.
Note: For multiple dosage strength, conduct linearity from 25% of the lowest strength to 150% of the highest strength. If standard and test concentrations are
different, establish linearity covering both the ranges.
5.1.1.1 Prepare a series of standard solutions ( Not less than six is recommended) in the above concentration range and scan using UV . Plot absorbance versus the actual concentration (µg/mL). Calculate the coefficient of correlation.
Note : Linearity range shall be altered based on the requirement.
5.1.2 Acceptance criteria:
The response is considered to be linear, if correlation coefficient (r) is not less than 0.999. In case of discrepancy investigate and explain the reason. Repeat if necessary and report the range in which it is linear.
5.2 SINK Condition for non compendial Dissolution Methods:
Note: Sink conditions need not be performed for compendia methods.
5.2.1 Determine solubility on three times the active drug substances in specified volume of dissolution medium in triplicate at room temperature (25°C), as per test method except RPM at 250.
5.2.2 Acceptance criteria:
NLT 1.5 times the active ingredient in one tablet or capsules or Oral suspension (of the highest dosage strength) should dissolve in the designated volume of the dissolution medium at room temperature.
5.2.3 If the above acceptance criteria is not met repeat point 4.2.1 to 4.2.2 at 37.5°C temperature.
5.2.4 If the acceptance criteria also not met at 37.5°C, also repeat point 4.2.1 to 4.2.2 with formulated blend at room temperature (25°C).
5.2.5 If the acceptance criteria is not met at 25°C for blend, repeat point 4.2.1 to 4.2.2 with formulated blend at 37.5°C temperature.
5.3 Method Precision:
5.3.1 Repeatability
5.3.1.1 Prepare six test preparations as per the test method on Tablets/Capsules/Oral suspension analyse and calculate % dissolution, average % dissolution of six unit dosages and RSD.
5.3.1.2 The method is considered to be “PRECISE”, if the % dissolution and average % dissolution meets the specification limit and relative standard deviation for % dissolution of six dosage units is not more than 5.0%.
5.3.1.3 If the method is used for over a range of dosage strength and test preparation issame for all dosage strength, then conduct method precision for highest and lowest dosage strength.
5.3.1.4 If the test preparation is different from strength to strength then conduct the method precision for all dosage strength.
5.3.1.5 If the method is used for a range of dosage types, (i.e. Tablets/ Capsules / Oral Suspension etc), then conduct method precision for each dosage type.
Note: If the precision fails perform validation using the product blend prepared by mixing the active ingredient with excipients as per the manufacturing formula.
5.4 Recovery / Accuracy:
5.4.1 Prepare homogeneous Tablet / Capsule blends having 25%, 50%, 75%, 100% and 150 % of the labeled amount of drug substances.
Note: Spiked levels of drug substances shall be altered based on the requirement.
5.4.2 Prepare blend by keeping average weight constant if the quantity of active ingredient is reduced by increasing the weight of placebo (or) prepare blends by keeping placebo constant.
5.4.3Prepare test solutions in triplicate for each spike level and analyse as per the test method. Calculate the % recovery and average % recovery.
Note: If % Recovery not meeting the acceptance criteria continue the dissolution at 200 RPM for 15minutes.
5.4.4 Acceptance criteria :
The method is considered to be “ACCURATE”, if the % recovery and average % recovery is not less than 95.0%.Note: In case of discrepancy investigate the reason, repeat the experiment if necessary and explain the reason for discrepancy.
5.5 Linearity of the Method:
5.5.1 Plot a graph with average “mg” added versus average “mg” found in recovery section (Refer 4.4) Evaluate the coefficient of correlation (r).
5.5.2 Acceptance criteria:
The coefficient of correlation (r) shall be NLT 0.999. If the correlation coefficient is less than 0.999, report the range in which the method is linear.
5.6 Specificity (Placebo interference)
5.6.1 Perform the dissolution as per the test method on weight of placebo in triplicate, equivalent to the amount present in weight of tablet powder / Capsule content. In case of Capsule use filled capsules filled with placebo.
5.6.2 Acceptance criteria:
Interference from placebo shall be NMT 2.0%.
5.7 Ruggedness:
5.7.1 System to system variability:
5.7.1.1 Conduct system to system variability study on two dissolution systems (of the same or different manufacturer). Perform the dissolution on six-unit dosage as per the test method.
5.7.1.2 Calculate the % dissolution, average % dissolution and RSD of six dosage units and Overall RSD for both the systems.
5.7.1.3 Acceptance criteria:
The method is considered to be rugged for system-to-system variability, if the % dissolution and average % dissolution, RSD meets the specification limit and Over all RSD should be not more than 5.0%, for both the systems.
5.7.2 Analyst to Analyst variability:
5.7.2.1 Conduct analyst to analyst variability study by two analysts at different days using the same UV-VIS Spectrophotometer and same dissolution system. Perform the dissolution on six dosage units as per the test method by using same sample.
5.7.2.2 Calculate the % dissolution, average % dissolution, RSD of six unit dosages and Overall RSD for both the analysts.
5.7.2.3 Acceptance criteria:
The method is considered to be rugged for analyst to analyst variability, if the % dissolution, average % dissolution, RSD meets the specification limit and over all RSD should be not more than 5.0% by both the analysts. Note: It is not considered necessary to study the variations (days, columns,
equipment and analysts) individually. The use of an experimental design (matrix) is encouraged
5.7.3 Bench top stability of standard and test preparations:
5.7.3.1 Establish the stability of standard and test solutions on bench top for a period of 2 days.
5.7.3.2 Prepare standard solution and test solutions in “duplicate” as per the test method and keep them on bench top. Scan standard and test solutions using UV-Visible spectrophotometer following the conditions described in the test method at initial, 1 day and 2 days. Calculate % dissolution of test solution and % assay of standard against a fresh standard each time.
Note: Keep potency of standard as initial Assay of the standard.
5.7.3.3 Acceptance criteria:
The solutions are considered to be “STABLE”, if the difference in % dissolution results from initial is not more than 2.0
5.7.3.4 If the solutions are found to be not stable for 1 day, measure the absorbance and calculate the results at shorter time intervals .e.g. At 1, 2, 3, hours interval and establish the period of time during which the solutions are stable.
5.7.4 Refrigerator stability of standard and test preparations:
5.7.4.1 Establish the stability of standard and test solutions in refrigerator for a period of about 2 days.
5.7.4.2 Prepare standard and test solutions in duplicate as per the test method and keep them in refrigerator. Scan the standard and test solutions using UV – Visible spectrophotometer following the conditions described in the test method at initial,1day and 2 days.
Note: Allow the samples to come to room temperature before measuring absorbance
5.7.4.3 Calculate % dissolution of test solution and % assay of standard against a fresh standard each time.
Note : Keep potency of standard as initial Assay of the standard
5.7.4.4 Acceptance criteria:
The solutions are considered to be “STABLE”, if the difference in % dissolution results from initial is not more than 2.0
5.7.4.5 If the solutions are found to be not stable for 1 day, measure the absorbance and calculate the results at shorter time intervals. E.g. At 1, 2, 3, hours interval and establish the period of time during which the solutions are stable.
5.8 Robustness:
5.8.1 Filter Validation:
5.8.1.1 To demonstrate that the filters do not affect the analytical results, validate different filters before use.
5.8.1.2 Prepare test solutions in triplicate and standard solution as per the test method without filtration. Centrifuge the portion of test solution and filter portions of test solutions through individual filters. Scan unfiltered standard solution & centrifuged and filtered test solutions using UV – Visible spectrophotometer under the test conditions, Calculate the % dissolution for centrifuged and filtered test solutions and difference of % dissolution between centrifuged and filtered samples.
5.8.1.3 Acceptance criteria:
The filter is considered to be acceptable, if the difference in % dissolution between centrifuged and filtered test solution is not more than 2.0
7.0 VERSION HISTORY


.JPG)












![Difference between Stability[Shelf life] Specification and Release Specification](https://4.bp.blogspot.com/-O3EpVMWcoKw/WxY6-6I4--I/AAAAAAAAB2s/KzC0FqUQtkMdw7VzT6oOR_8vbZO6EJc-ACK4BGAYYCw/w680/nth.png)
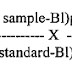
{kind=link}
0 Comments