
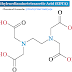
Elemental impurities in drug products may arise from several sources; they may be residual catalysts that were added intentionally in synthesis or may be present as impurities (e.g., through interactions with processing equipment or container/closure systems or by being present in components of the drug product). Because elemental impurities do not provide any therapeutic benefit to the patient, their levels in the drug product should be controlled within acceptable limits. There are three parts of this guideline: the evaluation of the toxicity data for potential elemental impurities; the establishment of a Permitted Daily Exposure (PDE) for each element of toxicological concern; and application of a risk-based approach to control elemental impurities in drug products. An applicant is not expected to tighten the limits based on process capability, provided that the elemental impurities in drug products do not exceed the PDEs. The PDEs established in this guideline are considered to be protective of public health for all patient populations. In some cases, lower levels of elemental impurities may be warranted when levels below toxicity thresholds have been shown to have an impact on other quality attributes of the drug product (e.g., element catalyzed degradation of drug substances). In addition, for elements with high PDEs, other limits may have to be considered from a pharmaceutical quality perspective and other guidelines should be consulted (e.g., ICH Q3A).

This guideline presents a process to assess and control elemental impurities in the drug product using the principles of risk management as described in ICH Q9. This process provides a platform for developing a risk-based control strategy to limit elemental impurities in the drug product.
2. SCOPE
The guideline applies to new finished drug products (as defined in ICH Q6A and Q6B) and new drug products containing existing drug substances. The drug products containing purified proteins and polypeptides (including proteins and polypeptides produced from recombinant or non-recombinant origins), their derivatives, and products of which they are components (e.g., conjugates) are within the scope of this guideline, as are drug products containing synthetically produced polypeptides, polynucleotides, and oligosaccharides.
This guideline does not apply to herbal products, radiopharmaceuticals, vaccines, cell metabolites, DNA products, allergenic extracts, cells, whole blood, cellular blood components or blood derivatives including plasma and plasma derivatives, dialysate solutions not intended for systemic circulation, and elements that are intentionally included in the drug product for therapeutic benefit. This guideline does not apply to products based on genes (gene therapy), cells (cell therapy) and tissue (tissue engineering). In some regions, these products are known as advanced therapy medicinal products. This guideline does not apply to drug products used during clinical research stages of development. As the commercial process is developed, the principles contained in this guideline can be useful in evaluating elemental impurities that may be present in a new drug product. Application of Q3



.JPG)











![Difference between Stability[Shelf life] Specification and Release Specification](https://4.bp.blogspot.com/-O3EpVMWcoKw/WxY6-6I4--I/AAAAAAAAB2s/KzC0FqUQtkMdw7VzT6oOR_8vbZO6EJc-ACK4BGAYYCw/w680/nth.png)
-Guideline for elemental impurities ){kind=link}
0 Comments